|
Ishida, H. (2014)
Essential function of the N-termini tails of the proteasome for the gating mechanism revealed
by molecular dynamics simulations , Proteins, 82, 1985-1999.
The proteasome is involved in the degradation of the majority of cellular proteins and plays an
important role in a wide variety of biological processes from protein quality control to DNA repair,
gene transcription, chromatin remodeling, cell-cycle control, signal transduction, antigen presentation,
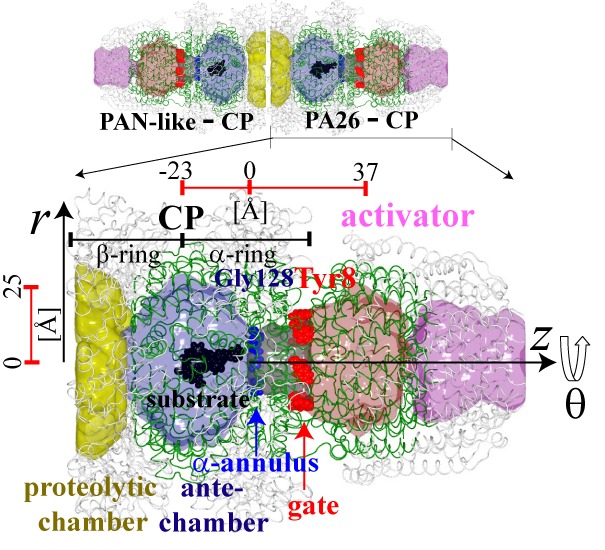
and so on. The 19S-CP-PA28 complex called a hybrid proteasome (Fig. 1)
is thought to be responsible for the efficient proteolysis of the substrate,
and to be involved in the immunological processing of intracellular antigens.
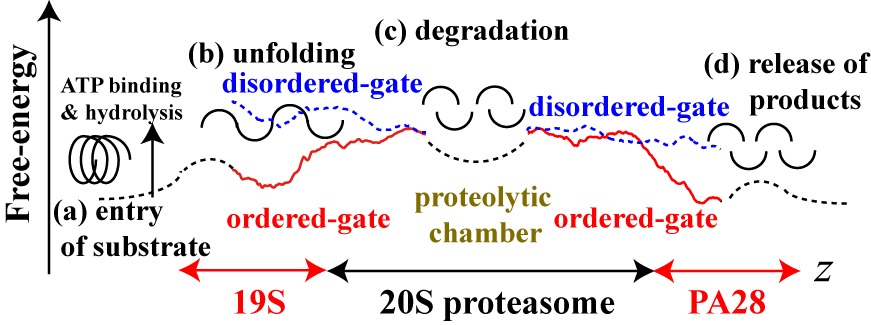
The free-energies of the translocation of a substrate moving through the 19S-CP-PA28
complex were estimated as shown in Figure 2. From the results,
a model of the entry of the substrate and the release of the products
by the hybrid proteasome has been proposed (Fig. 2).
|