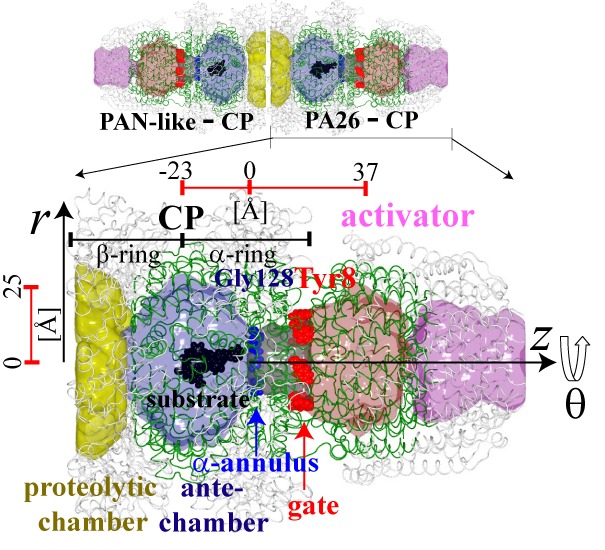
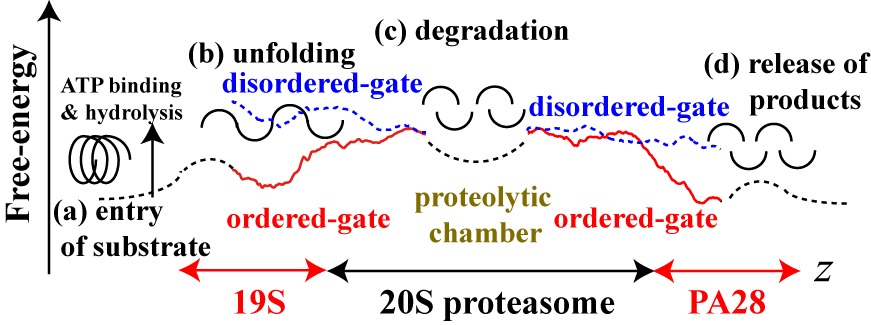
|
Ikebe, J., Sakuraba, S. and Kono, H. (2014)
Adaptive lambda square dynamics simulation: An efficient conformational sampling method for biomolecules
J Comput. Chem., 35, 39-50.
蛋白質や DNAは、それぞれアミノ酸やヌクレオチドが鎖状に連なった生体分子です。
この鎖状分子は、低い自由エネルギーを持つ特定の立体構造に折り畳まれることに
よってその機能を発揮します。分子動力学シミュレーションで生体分子の自由エネルギー地形を
再現することは、生物学の大きな目標のひとつです。しかし、従来のシミュレーション手法で
これを実現するためには莫大な時間を要します。そこで私たちは従来の手法よりも高速に
生体高分子の自由エネルギー地形を再現することのできる手法、
Adaptive Lambda Square Dynamics (ALSD) 法を開発しました。
ALSDは、生体分子のポテンシャルエネルギー変化を促進させることによって、
様々な立体構造を高速に探索することができます。私たちは、
ポリリジン9残基ペプチドのテスト計算を行うことによって、
ALSD は従来手法(McMD)よりも高速に、このペプチドの自由エネルギー地形を
再現できることを確認しました。
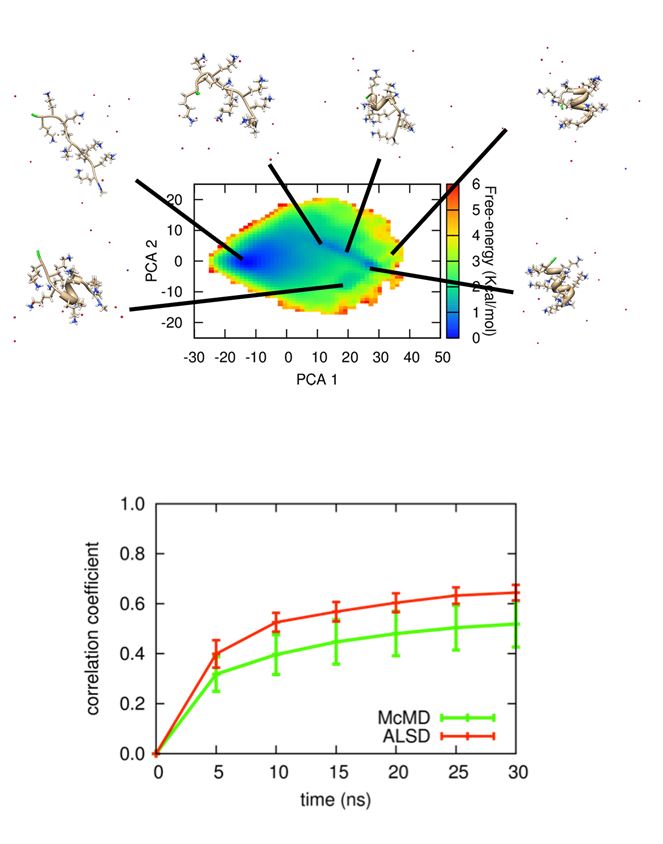
(左上図)主成分解析(PCA)法によって描かれたポリリジン9残基ペプチドの自由エネルギー地形と代表構造。
(左下図)十分に長時間のシミュレーションから得られた上図の自由エネルギー地形と、
短時間シミュレーションから得られた自由エネルギー地形の間の相関係数。
|